6 代谢模型
6.1 Genome-scale Models
大多数自然微生物系统都是在具有时空变化的环境中进化而来的。理解这种复杂系统的一个主要局限是缺乏数学建模框架,无法将单个物种的基因组和环境的时空变化与系统行为联系起来。时空通量平衡分析(SFBA)将通量平衡分析(FBA)扩展到考虑环境的时空变化,是微生物代谢基因组尺度重建的新兴领域。SFBA 预计是微生物代谢建模的下一个前沿领域,未来的方法开发和系统应用将迅速增加。
6.1.1 通量平衡分析及其动态扩展
通量平衡分析法(FBA)是一种有限描述全细胞代谢的建模方法,该方法的基础是代谢物质量守恒和反应化学计量学。因为化学计量学模型描述了通过纳入基因组中注释的生化反应来构建的代谢通路,由此产生的模型通常被称为基因组尺度代谢(GSM)模型。
给定化学计量矩阵和一组指定的可用养分吸收率,目标是求解未知细胞内反应速率(即细胞内通量)和代谢副产物分泌速率(即分泌通量)的 GSM。由于比起质量平衡方程,化学计量方程总是包含更多的未知通量,因此 GSM 有无限多的解,通量分布无法唯一确定。为了克服这一局限性,假定细胞新陈代谢受到调节,以实现某种细胞目标。最常见的目标是最大限度地生长,由此就将生物代谢物(如 RNA、DNA、蛋白质、碳水化合物等)的化学计量方程结合了起来,产生一个线性规划(LP)问题,通过有效求解,可预测细胞内通量分布、吸收和分泌(交换)通量以及生长率( Figure 6.1 )。
6.1.2 SFBA 问题及最新进展
FBA 所依据的假设是细胞内代谢和细胞外环境都是时间不变的。动态 FBA(DFBA)则通过扩展取消了细胞外环境的稳态假设1。DFBA 模型是在细胞迅速适应环境变化的假设条件下,将细胞内代谢的 GSM 与生长限制性营养物质吸收率的动力学表达式以及细胞生物量、限制性营养物质和分泌的代谢副产物的动态质量平衡方程相结合而形成的( Figure 6.2 )。DFBA 的一个主要优点是:模型输出包括生物量和细胞外代谢物浓度,而不像 FBA 那样只包括生长速率和交换通量。此外,对细胞外浓度和细胞内通量的预测具有时间分辨率。
尽管 DFBA 考虑了细胞外动态对细胞内代谢的影响,但该方法是基于细胞外环境混合良好且空间均匀的假设。同时考虑空间和时间变化的数学模型通常称为时空模型。我们把包括环境空间异质性的 DFBA 扩展称为时空 FBA(Spatiotemporal FBA,SFBA)。
SFBA 中用随时间和空间坐标变化的偏微分方程(Partial Differential Equations,PDEs)取代了 DFBA 中的随时间变化的 ODEs2。偏微分方程表示生物量、代谢物和可能的其他化学物种浓度的细胞外质量平衡方程,并说明引起空间变化的传输机制,通常包括代谢物扩散和液相/气相对流。必须在空间域的边界施加边界条件,以确保 PDE 得到良好的拟合。与 DFBA 一样,GSM 和细胞外质量平衡方程通过养分吸收动力学联系在一起。
营养物质和代谢产物在生物膜不同位置存在异质性。而且,由于细胞的生长和/或死亡,空间异质性可能会随着时间的变化。为便于说明,考虑厚度固定为 \(L\) 的单一物种生物膜中,底部( \(z=0\) )有单一限制生长的营养物质,顶部( \(z=L\) )有单一合成的副产品排出。假设空间变化只发生在生物膜的轴向 \(z\),描述生物膜扩散过程的 PDE 可写成以下形式:
\[ \begin{array}{l l l l} \frac {\partial X(z,t)}{\partial t} = \mu X \qquad & X(z, 0) = X_I \\ \frac {\partial S(z, t)}{\partial t} = \upsilon_S X + D_S \frac {\partial^2S}{\partial z^2} \qquad & S(0,t) = S_0 \quad & \frac {\partial S(L,t)}{\partial z} = 0 \quad & S(z,0) = S_I \\ \frac {\partial P(z,t)}{\partial t} = v_P X + D_P \frac {\partial ^2 P}{\partial z^2} \qquad & \frac{\partial P(0,5)}{\partial z}=0 \quad & P(z,t)=0 \quad & P(z,0)=P_I \end{array} \]
这里的 \(X(z,t)\)、\(S(z,t)\) 和 \(P(z,t)\) 分别表示在 \(z\) 处和时间 \(t\) 的生物量、底物浓度和副产物浓度。通过求解 GSM 可以得到生长率 \(μ\)、底物吸收率 \(v_S\) 和副产物合成率 \(v_P\)。基质和副产物分别以高效扩散系数 \(D_S\) 和 \(D_P\) 在生物膜中扩散,而生物质被假定为无运动性。第二栏列出了生物膜底部的边界条件,其假设条件是:基质的浓度为 \(S0\),生物质和副产物不会穿过该边界。第三列生物膜顶部的边界条件是基于以下假设:生物质和基质不会流过该边界,而副产品的去除率足够高,以至于边界浓度为零。最后,第四栏中的初始条件是基于生物膜在 \(t=0\) 时空间均匀的假设。
SFBA 模型由描述细胞外环境的 PDE 和描述细胞内代谢的 LP 组成。纯物种系统只有一个 LP ,而多物种系统则需要求解每个物种的 LP。由于没有直接求解这种混合 PDE-LP 模型的计算方法,因此必须以某种方式近似 PDE 的传输行为,以生成可求解的模型。Figure 6.3
6.1.3 SFBA 案例研究
6.1.4 COMETS 模拟合成群落演化
2014 年发表在 Cell Reports 上的研究开发了 COMETS 的时空建模框架3,并将其用于研究二物种和三物种合成群落的涌现性行为(Harcombe et al. 2014)。COMETS 定义了一个二维网格(2D lattice),网格中的每个方格(box)代表一个不同的空间位置。假定每个物种都有可能根据相同的吸收动力学消耗可用碳源,则可使用参与物种的 GSM 解决每个方格的独立 DFBA 问题。采用了一种顺序求解策略,即先求解物种 LP,以生成本地生长速率和通量,然后在一个固定的时间步长内将细胞外 ODE 与这些恒定的 LP 求解进行整合,以生成本地生物量和代谢物浓度。在求解 LP 开始下一次迭代之前,在同一时间步长内求解二维扩散方程,以允许物种细胞质量和代谢物在方格之间扩散。
研究使用 COMETS 模拟了由沙门氏菌和大肠杆菌组成的合成群落中的特定代谢物互养过程。该时空模型能够重现实验结果,即两种生物稳定共存,并收敛到大肠杆菌种群比例占 79% 的结局,且与初始比例无关。模拟由沙门氏菌、大肠杆菌和外生甲基杆菌组成的三物种合成群落也得到了类似的结果。双物种系统用于研究空间结构的影响。该模型正确地预测出,随着两个物种接种的距离越远,群落的生长速度就会越慢,这是由于交叉喂养的代谢物的扩散限制所致。此外,该模型还再现了实验结果,即在沙门氏菌和大肠杆菌种群之间放置第二个基因工程沙门氏菌菌落会促进群落生长,但放置不需要大肠杆菌交叉喂养代谢物的野生型沙门氏菌则会降低群落生长。
6.1.5 DFBAlab 求解乙醇生物发酵问题
来源:(Chen et al. 2015)
预测了厌氧梭菌在垂直气泡塔反应器中将合成气(\(\ce{CO}\)、\(\ce{H2}\))转化为乙醇的情况。合成气和液体介质进料流被引入气泡塔底部,并以不同的速度沿气泡塔向上流动,由于细胞生长和气体耗竭,产生了较大的空间梯度。代谢副产物乙醇和乙酸酯在从塔顶流出的液相流中回收。时空代谢模型由已发表的 C. ljungdahlii GSM [69]、\(\ce{CO}\) 和 \(\ce{H2}\) 的 Michaelis-Menten 吸收动力学以及 C. ljungdahlii 生物质、液相 \(\ce{CO}\)、\(\ce{H2}\)、乙醇和乙酸盐以及气相 \(\ce{CO}\) 和 \(\ce{H2}\) 的反应对流型 PDEs 组成。这些 PDE 被离散化为 100 个空间结点,并在每个结点使用六个 LP 进行词法优化,以确保交换通量的唯一性。由 900 个时间 ODE 和 600 个 LP 组成的大型离散模型在 MATLAB(MathWorks)中使用 DFBAlab(DFBA 实验室)工具进行了有效求解( Figure 6.3 )。
针对各种色谱柱操作条件和养分吸收参数进行的动态模拟,得出了与现有实验一致的色谱柱行为和乙醇生产瓶颈预测,这些实验包括:(1)典型的富含 \(\ce{CO}\) 的合成气会在塔的上部由于 \(\ce{H2}\) 耗竭而产生大量醋酸盐,这表明增加合成气进料中的 \(\ce{H2}\) 可能是有益的;(2)高效的气液传质是实现高乙醇产量和高转化率的关键,这表明需要继续开发先进的气泡塔设计,以实现极高的气液传质速率;以及 (3)提高 \(\ce{H2}\) 的吸收率可大幅提高乙醇滴度和乙醇-醋酸盐比率,这表明通过改良 C. ljungdahlii 的工程设计提高 \(\ce{H2}\) 的吸收率可实现高乙醇产量和高转化率。
6.1.6 未来可能的研究方向
基因组尺度模型是生物体已知或推断的代谢网络的数学表示[72]。利用基因组尺度模型进行模拟,可预测生物体如何利用环境中的特定资源进行生长,以及由于分泌、细胞外降解或泄漏而产生的任何新的代谢物(图 2)。因此,利用多个生物体的模型进行模拟(通过将数字生命体视为独立的组件来进行),可以对资源竞争、生态位划分或代谢交叉喂养进行定量预测,并已应用于多种系统,包括人类肠道微生物群73、74、75。(Pacheco and Vorholt 2023)
在活体植物中,将基因组尺度模型的预测结果与合成群落实验相结合,为了解更复杂的代谢相互作用的复杂性提供了一条特别有前途的途径[27]。例如,这种综合方法最近揭示了从杨树根瘤中分离出来的细菌群落稳定性的代谢因素[76]。此外,在空间范围内模拟代谢相互作用的新方法 77、78 可以预测多物种在环境中的相互作用,这种环境近似于与植物相关的微环境 79、80、81 中与空间和时间相关的资源条件。高通量培养技术[82]、代谢组学技术[83]和可视化技术[84]的进步也有望提高更复杂群落尺度下相互作用预测的准确性。(Pacheco and Vorholt 2023)
值得注意的是,Borer 等人最近利用了这一技术组合,他们使用经过实验验证的四种特征良好菌株的基因组尺度模型来预测土壤细菌之间的群落组装和代谢交换(Borer, Kleyer, and Or 2022)。这项研究进一步突出了基因组尺度模型描述空间分辨环境中相互作用机制的能力,表明预测复杂环境中群落生态学的潜力越来越大。
6.2 COMETS
COMETS(Computation Of Microbial Ecosystems in Time and Space) is a software platform for performing computer simulations of spatially structured microbial communities. It is based on stoichiometric modeling of the genome-scale metabolic network of individual microbial species using dynamic flux balance analysis, and on a discrete approximation of diffusion. For more information, see Harcombe et al., Metabolic Resource Allocation in Individual Microbes Determines Ecosystem Interactions and Spatial Dynamics, Cell Reports, 2014.
6.2.1 COMETS 软件特性
COMETS 的核心是通过迭代方式最大化目标反应(通常是生物量生产)来模拟微生物种群的生长。一个或多个物种的初始生物量(由其代谢模型定义)被播种到含有一系列指定营养物质的环境中。在每次迭代中,生物量和环境都会根据 FBA 预测进行更新。
6.2.1.1 空间能力
COMETS 能够模拟空间结构环境中的微生物生长。这是通过将模拟空间(“世界”)划分为由较小空间组成的 “网格”来实现的。在每个空间内,生长被认为是充分混合的。随着模拟的进行,营养物质和生物量都可以传播到连续的空间。
模拟二维和三维 “世界”
除了混合良好的条件外,COMETS 还能模拟二维和三维空间结构环境。例如,可以模拟培养皿等二维表面上的菌落生长,或肿瘤、三维基质中的细菌菌落等三维结构。
生物质在空间的扩散和对流传播
在具有空间结构的模拟中,可以实现不同的生物质传播模式。扩散模式模拟的是自由游动细菌的传播,而对流模式模拟的是细菌相互推动的传播。这两种传播模式可以结合使用。
取决于基质的营养物质和生物量传播
营养物质的扩散性和生物质的传播特性取决于琼脂密度、细胞基质摩擦系数等基质特性。
边界条件
有两种边界条件。固定值和固定源或汇速率。
6.2.1.2 生物功能
COMETS 具有许多有趣的生物功能,可用于完善和改进化学计量模型的预测,以及模拟不同类型的生物现实条件。
微生物生长的滞后期
滞后期是模拟菌落生长激活的模型。
连续(恒温箱)和批量生长模式
在恒温箱模式下,用户可控制营养液的补充速度。在批处理模式下,用户可控制稀释和频率。
多物种群落模拟
可在物种空间分布重叠或不重叠的情况下,或在混合良好的条件下,对两个或多个物种(最多可达数百个)进行模拟。
解析性 dFBA
通常,任何代谢模型都有多个最优解。在这些方案中进行选择的一种方法是假设细胞将通过代谢网络的总通量最小化。为此,解析式 FBA 首先优化目标函数,如生长。然后进行第二次优化,将生长量固定在之前获得的最佳水平,并使通过网络的总通量最小化。
细胞死亡
我们采用了一个简单的细胞死亡模型,并为每个物种分配了死亡率。
中性种群漂移
人口噪声的存在会导致模拟中不同物种丰度的随机变化。这在批量增长模式中尤其有用,因为稀释瓶颈会对增长中的种群产生重大影响。
进化过程
Comets 支持进化模拟,包括模拟过程中的随机变异和漂移。目前,唯一可用的突变是反应删除。
6.2.1.3 计算能力
COMETS 软件使用 JAVA 语言实现。因此,它具有很强的可移植性,不受操作系统的影响。COMETS 具有以下模拟功能。
图形用户界面 (GUI)
除命令行外,COMETS 仿真还可通过图形用户界面(包括可视化工具)运行。
并行 dFBA
在多 CPU 系统中以多线程进程方式运行,可获得更高的计算性能。
MATLAB 工具箱
MATLAB 工具箱,用于以编程方式修改 COMETS 的输入文件。
Python 工具箱
Python 工具箱,用于以编程方式修改 COMETS 的输入文件。
6.3 使用 DFBAlab
Write a dFBA model
Dynamic flux balance analysis is an extension of FBA that has the advantage that it enables analysis of interactions between the metabolism and the environment. The structure of the dFBA model that is assumed here has the following elements:
- A flux balance model of each of the microorganisms.
- A list of external metabolites.
- Description of the interaction between FBA models through uptake and production of external metabolites.
- The dynamic equations that are integrated by DFBAlab.
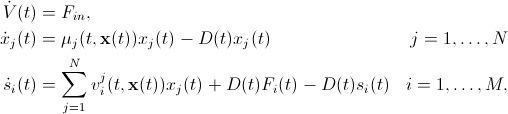
where \(N\) is the number of biological species and \(M\) is the number of extracellular chemical species. The variable \(x_j\) is the biomass concentration of the \(j^{th}\) metabolism, \(s_i\) is the extracellular concentration of the \(i^{th}\) chemical species, \(F_{in} = \sum_{i=1}^{M}F_{i} (t)\) with \(F_i\) is the feed rate of chemical species \(i\), \(D=\sum_{i=1}^{M}F_i /V\) is the dilution rate, \(v_i^j\) is the uptake or production rate of chemical species \(i\) due to species \(j\) and \(V\) is the volume of the bio-reactor. If the function \(v_i^j\) describes the production rate of the \(i^{th}\) external metabolite by the \(j^{th}\) metabolism, then its value can be determined by secondary optimization for the production rate.
Setup the simulation
The simulation parameters are determined by the batch conditions of the experiments. A minimal set of parameter consists of
- The (maximal) batch time.
- The initial conditions for the biomass, substrates and total volume.
- Definition of the external control inputs such as feed rate and composition and other environmental changes that are should be implemented during the batch.
- Optional definition of minimal growth conditions to determine when the simulation should stop prior to reaching the final batch time.
6.4 Community Simulator
Citation: Marsland R, Cui W, Goldford J, Mehta P (2020) The Community Simulator: A Python package for microbial ecology. PLoS ONE 15(3): e0230430. https://doi.org/10.1371/journal.pone.0230430
Community Simulator is a Python package called Community Simulator for simulating microbial population dynamics in a reproducible, transparent and scalable way.
The Community Simulator includes five major elements: tools for preparing the initial states and environmental conditions for a set of samples, automatic generation of dynamical equations based on a dictionary of modeling assumptions, random parameter sampling with tunable levels of metabolic and taxonomic structure, parallel integration of the dynamical equations, and support for metacommunity dynamics with migration between samples.
This package is designed for simulating batch culture experiments on complex microbial communities. The architecture is based on the standard protocol for these experiments:
Add media to each of the wells in a 96-well plate. It could be the same media for all wells, or different media for each well, depending on what the experimenter is trying to test.
Add a small sample of an existing bacterial culture to each of the wells. Again, these could be the same for all wells, or different initial conditions could be tested in parallel.
Let the bacteria grow while stirring or shaking for some fixed time T.
Pipette a small fraction of each of the wells into a well on a new plate, with fresh media (added according to the same protocol as in step 1).
Repeat the previous two steps as many times as desired.
Communities can also be run in chemostat mode, where nutrients are continually supplied and populations continuously diluted.